



ICU Management & Practice, Volume 22 - Issue 2, 2022
 PRINT OPTIMISED
PRINT OPTIMISED
Introduction
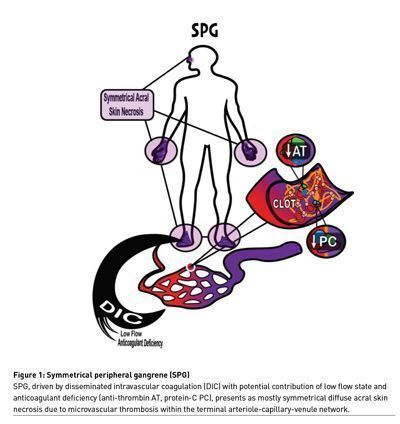
Symmetrical peripheral gangrene (SPG) refers to the development of tissue necrosis in an acral distribution without vasculitis and often without loss of arterial pulses. It is a rare but devastating clinical syndrome associated with significant morbidity and mortality, often leading to amputation of digits or limbs in patients who survive.
SPG is most commonly seen with sepsis, but has been described in a wide variety of clinical conditions. A typical presentation would be in a patient with septic shock and disseminated intravascular coagulation (DIC) who initially develops skin mottling temporally related to a sudden and progressive drop in platelet count. Evidence of diffuse acral cyanosis and tissue loss then follow.
The pathophysiology is incompletely understood but thought to result from a DIC driven microvascular thrombosis often paired with impaired perfusion, and relative deficiency in anti-coagulant factors such as protein C and antithrombin secondary to impaired production and consumption (Warkentin and Ning 2021). Ischaemic hepatic injury as a source for the reduction in the aforementioned liver produced anti-coagulant factors is thought to play a significant role (Warkentin and Ning 2021). DIC is likely the final common pathway of SPG. SPG has in fact been described as the cutaneous marker of DIC (Sharma et al. 2004).
While vasopressor administration has been associated with SPG, a causative or contributory role has never been established. One reason that SPG remains poorly understood is the rarity of its presentation. Research has been limited to animal models, case reports, and small single-centre case series of patients with a wide spectrum of clinical illness. Modifiable risk factors and effective treatment options remain poorly defined.
In this article, we briefly review the pathophysiology, potential treatments, and areas for future research.
Pathophysiology
SPG has been described as its own clinical entity, but exists along a spectrum of other related thrombotic disorders that lead to tissue necrosis. These include purpura fulminans, venous limb ischaemia, warfarin skin necrosis, and heparin induced thrombocytopenia (Warkentin 2015). Purpura fulminans is most closely associated, and is the term used to describe the presence of both acral and non-acral gangrene in a relatively symmetrical distribution.
All of these disorders involve coagulopathy with coagulation factor consumption and anti-coagulant insufficiency resulting in thrombosis and tissue gangrene (Warkentin 2015). Yet, while venous limb ischaemia, warfarin skin necrosis, and heparin induced thrombocytopenia have well recognised precipitants – deep vein thrombosis, coumadin administration, and heparin-platelet factor 4 antibody development, respectively – conditions precipitating SPG are more varied.
SPG was first described by J Hutchinson in a 37-year-old male with shock (Hutchinson 1891). It has since been described in association with sepsis, COVID-19 infection (Sil et al. 2022), myocardial infarction (Caserta et al. 1956), pancreatitis (Liao et al. 2015), trauma (Tan et al. 2018), and envenomation (Shastri et al. 2014), among others. While SPG is most commonly reported in patients with hypotension, it has also been reported in individuals without overt hypotension (Kurup and Simpson 2019).
The shared common pathway in SPG is thought to be a triad of 1) end organ shock, 2) disseminated intravascular coagulation, and 3) depletion of natural anticoagulants through consumption and impaired production (Warkentin and Ning 2021) leading to small vessel fibrin thrombosis (Robboy et al. 1973) and tissue necrosis. Liver dysfunction due to ischaemic hepatitis is observed in many cases (Warkentin and Ning 2021), but dilution of anticoagulants may also occur through colloid volume expansion (Warkentin et al. 2020).
Vasopressors are often cited as having a significant role in developing SPG (Hayes et al. 1992; Ruffin et al. 2018). However, such causality has never been established. Several observations argue against it. First, the majority of patients on high dose vasopressors never develop SPG. Second, there may be a significant time delay between vasopressor initiation and the onset of distal tissue ischaemia (Warkentin and Ning 2021). Third, there are patients who develop SPG prior to ever receiving vasopressors. Ghosh et al. (2010) reported a case series of 14 consecutive patients presenting with SPG. None of them received vasopressors immediately prior to or during the development of tissue gangrene.
It is possible that vasopressors exacerbate SPG. However, the evidence for this is very weak. Kwon et al. (2018) performed a small retrospective matched case-control study comparing 36 patients with SPG with 42 controls. They found that weight adjusted mean dose of dopamine and the weight adjusted peak doses of norepinephrine, dopamine, and epinephrine correlated with SPG. However, the SPG group was more haemodynamically unstable, thrombocytopenic, and had lower Glasgow Coma Scale (GCS) scores than the control group (Kwon et al. 2018). Coagulation markers were also not reported. Many other case reports attribute SPG to vasopressor use. But association does not mean causality. Of note, vasopressors have not received the same level of attention as a contributing factor in purpura fulminans.
Proposed Treatment Options
Various treatment strategies have been described for SPG. Most of these treatments are based on sound theory, but lack robust evidence to support their efficacy or safety. Proposed interventions can be broadly classified in terms of 1) anti-coagulation, 2) replacement of natural anti-coagulant factors, 3) improving circulation, and 4) reduction in systemic inflammation.
Anti-coagulation
Several case reports describe using systemic anti-coagulation to treat SPG (Tripathy and Rath 2010; Kurup and Simpson 2019). However, the role of this remains uncertain and must be weighed against bleeding risk in the setting of thrombocytopenia (Warkentin and Ning 2021). This is in line with variable society recommendations for anti-coagulation in DIC without bleeding (Wada et al. 2014).
If systemic heparin anti-coagulation is used, monitoring and dosing requirements may be affected by DIC related elevations in partial thromboplastin time and reductions in antithrombin, respectively (Warkentin and Ning 2021).
Replacement of anti-coagulant factors
Replacement of anti-coagulant factors has also been proposed as a treatment strategy for SPG. This is largely based on case series suggesting possible benefit of administering protein C concentrate to patients with purpura fulminans from sepsis (Rintala et al. 1998; Schellongowski et al. 2006) or congenital protein C deficiency (Manco-Johnson et al. 2016).
Antithrombin III may also be depleted in patients with SPG and severe sepsis in general. However, antithrombin III repletion has not been shown to reduce mortality or the incidence of new organ dysfunction in patients with severe sepsis, and may increase the risk of bleeding (Warren et al. 2001).
Improving circulation
Various methods to improve circulation in the microvascular bed have been described. These include administering systemic or topical vasodilators (e.g., topical nitroglycerin ointment), alpha receptor or sympathetic blockade, volume expansion, and reduction in vasopressor dosing (Willis et al. 2001; Foead et al. 2018; Kurup and Simpson 2019). None of these therapies have proven efficacy. Since in septic shock associated DIC vasopressors are primarily used to combat mediator induced systemic vasodilation, the addition of vasodilator therapy would seem counterintuitive. In addition, several of these interventions have the potential to further worsen haemodynamics for patients already in shock. Colloid volume expansion may also worsen natural anti-coagulant deficiency (Warkentin et al. 2020).
Reducing systemic inflammation
Limited case reports also cite haemofiltration (Smith et al. 1997) or haemoabsorption (Uncu Ulu et al. 2021) as helping to reduce or reverse peripheral tissue changes of SPG. These are based on the idea that such methods cause reductions in circulating inflammatory mediators that may be contributing to the syndrome. However, such methods have not shown improvements in mortality or organ dysfunction in a general septic shock population where inflammatory mediators are thought to be a driver of organ dysfunction (Dellinger et al. 2018).
Ultimately, early identification and aggressive treatment of the underlying cause should remain the management priority in all patients with SPG.
Future
Research on SPG is limited by the infrequent occurrence of this syndrome and the yet to be defined clinical markers that precede visual ischaemic changes. Cases may also go unreported due to rapidity of progression and when SPG is seen as an unavoidable consequence of multi-organ dysfunction or attributed to vasopressor administration. Research is likely to remain limited to sporadic case reports and small case series that often combine multiple treatment strategies, describe subjective clinical improvement without control comparisons, are subject to publication bias, and occur over prolonged periods of time when clinical practices may have changed.
Registry data collection may offer a solution. A registry would allow for centralised collection of standardised data to improve understanding of the modifiable risk factors, treatments, and natural course of SPG. More informed prospective studies could then follow. Brunkhorst and Patchev (2014) recognised this in creating The Sepsis-associated Purpura Fulminans International Registry – Europe. While this registry was ultimately closed due to challenges with funding and enrolment, it highlights the need to better understand this important clinical syndrome.
Conflict of interest
None.
References:
Brunkhorst F, Patchev V (2014) Sepsis-associated Purpura Fulminans International Registry--Europe (SAPFIRE). Medizinische Klinik, Intensivmedizin und Notfallmedizin. 109(8):591-595.
Caserta S, Metz R, Anton M (1956) Symmetrical Peripheral Gangrene in Myocardial Infarction: Report of a Case. New England Journal of Medicine. 254(12):568-570.
Dellinger RP, Bagshaw SM, Antonelli M et al. (2018) Effect of targeted polymyxin B hemoperfusion on 28-day mortality in patients with septic shock and elevated endotoxin level: the EUPHRATES randomized clinical trial. JAMA. 320(14):1455-1463.
Foead AI, Mathialagan A, Varadarajan R, Larvin M (2018) Management of symmetrical peripheral gangrene. Indian journal of critical care medicine. 22(12):870.
Ghosh S, Bandyopadhyay D, Ghosh A (2010) Symmetrical peripheral gangrene: a prospective study of 14 consecutive cases in a tertiary-care hospital in eastern India. Journal of the European Academy of Dermatology and Venereology. 24(2):214-218.
Hayes M, Yau E, Hinds C, Watson J (1992) Symmetrical peripheral gangrene: association with noradrenaline administration. Intensive care medicine. 18(7):433-436.
Hutchinson J (1891) Severe symmetrical gangrene of the extremities. Br Med J. 2:8-9.
Kurup V, Simpson RS (2019) Sepsis-induced digital ischaemia in a professional pianist, in the absence of vasopressors. BMJ Case Reports CP. 12(10):e229659.
Kwon JW, Hong MK, Park BY (2018) Risk factors of vasopressor-induced symmetrical peripheral gangrene. Annals of plastic surgery. 80(6):622.
Liao CY, Huang SC, Lin CH et al. (2015) Successful resolution of symmetrical peripheral gangrene after severe acute pancreatitis: a case report. Journal of medical case reports. 9(1):1-4.
Manco-Johnson MJ, Bomgaars L, Palascak J et al. (2016) Efficacy and safety of protein C concentrate to treat purpura fulminans and thromboembolic events in severe congenital protein C deficiency. Thrombosis and Haemostasis. 116(07):58-68.
Rintala E, Seppala OP, Kotilainen P et al. (1998) Protein C in the treatment of coagulopathy in meningococcal disease." Critical care medicine. 26(5):965-968.
Robboy SJ, Mihm MC, Colman RW, Minna JD (1973) The skin in disseminated intravascular coagulation: Prospective analysis of thirty-six cases. British Journal of Dermatology. 88(3):221-229.
Ruffin N, Vasa CV, Breakstone S, Axman W (2018) Symmetrical peripheral gangrene of bilateral feet and unilateral hand after administration of vasopressors during septic shock. Case Reports 2018. bcr-2017-223602.
Schellongowski P, Bauer E, Holzinger U et al. (2006) Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin®). Vox sanguinis. 90(4):294-301.
Sharma B, Kabra S, Gupta B (2004) Symmetrical peripheral gangrene. Tropical doctor. 34(1):2-4.
Shastri M, Parikh M, Patel D et al. (2014) Symmetrical peripheral gangrene following snake bite. Journal of Clinical and Diagnostic Research. 8(9):MD03.
Sil A, Chakraborty U, Chandra A, Biswas SK (2022) COVID-19 associated symmetrical peripheral gangrene: A case series. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 16(1):102356.
Smith O, White B, Vaughan D et al. (1997) Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. The Lancet. 350(9091):1590-1593.
Tan JH, Mohamad Y, Tan CLH et al. (2018) Concurrence of symmetrical peripheral gangrene and venous limb gangrene following polytrauma: a case report. Journal of medical case reports. 12(1):1-5.
Tripathy S, Rath B (2010) Symmetric peripheral gangrene: Catch it early! Journal of Emergencies, Trauma and Shock. 3(2):189.
Uncu Ulu B, Yiğenoğlu TN, Hacıbekiroğlu T et al. (2021) Recovery of symmetrical peripheral gangrene of limbs in a patient after performing hemoadsorption in septic shock. Journal of clinical apheresis. 36(4):649-653.
Wada H, Matsumoto T, Yamashita Y (2014) Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. Journal of Intensive Care. 2(1):1-8.
Warkentin TE (2015) Ischemic limb gangrene with pulses. New England Journal of Medicine. 373(7):642-655.
Warkentin TE, Ning S (2021) Symmetrical peripheral gangrene in critical illness. Transfusion and Apheresis Science. 60(2):103094.
Warkentin TE, Ning S, Lim W (2020) Colloid transfusion, natural anticoagulant depletion, and symmetric peripheral gangrene. New England Journal of Medicine. 383(16):1592-1594.
Warren BL, Eid A, Singer P et al. (2001) High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 286(15):1869-1878.
Willis TMS, Hopp RJ, Romero JR, Larsen PD (2001) The protective effect of brachial plexus palsy in purpura fulminans. Pediatric neurology. 24(5):379-381.















